Tutorial: countsplitting and scran
scran_tutorial.RmdBefore using this tutorial, we recommend that you read through our introductory tutorial to understand our method in a simple example with simulated data.
In this tutorial, we use a real dataset from Elorbany et al. (2022). The dataset contains 10,000 cells collected over 15 days of a directed differentiation protocol from induced pluripotent stem cells (IPSC) to cardiomyocytes (cm).
Overview
After loading the necessary software and performing count splitting, in this tutorial we carry out the following steps.
Preprocessing: We preprocess the training data using the preprocessing steps suggested by the scran tutorial.
Clustering: We cluster the cells using
scranusing the preprocessed training data.Differential Expression Analysis: Using the test data, we test to see which genes are associated with the estimated clusters. We do this both “by hand” using Poisson GLMs as well as using the built-in methods from the
scranpackage.
Install scran
If you don’t already have scran, you will need to
run:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("scran")Next, you should load the package, along with others that we will use in this tutorial.
Load the data and perform count splitting
This data is included in the countsplit_tutorials
package as a SingleCellExperiment object, so it is simple
to load.
data(cm)
cm## class: SingleCellExperiment
## dim: 21971 10000
## metadata(0):
## assays(1): counts
## rownames(21971): AL627309.1 AL627309.6 ... AC136352.4 AC007325.4
## rowData names(0):
## colnames(10000): E1_E1CD3col4_CATTTGTGCTTG E1_E1CD3col2_AGAATAAGTCAC
## ... E1_E1CD1col5_GTTACGCTAGTG E1_E1CD2col4_CCGCACAAGATC
## colData names(26): orig.ident nCount_RNA ... pseudotime ident
## reducedDimNames(0):
## mainExpName: NULL
## altExpNames(0):The main item in cm that we care about is the counts
matrix, which contains 21,971 genes and 10000 cells. We can view a small
subset of it now.
dim(counts(cm))
## [1] 21971 10000
counts(cm)[1:10,1:10]
## 10 x 10 sparse Matrix of class "dgCMatrix"
## [[ suppressing 10 column names 'E1_E1CD3col4_CATTTGTGCTTG', 'E1_E1CD3col2_AGAATAAGTCAC', 'E2_E2CD2col5_ATGAATGATGAA' ... ]]
##
## AL627309.1 . . . . . . . . . .
## AL627309.6 . . . . . . . . . .
## AL627309.5 . . . . . . . . . .
## AL669831.3 . . . . . . . . . .
## MTND1P23 1 12 . . 3 1 . . 4 .
## MTND2P28 6 3 . . . . 2 1 . .
## MTCO1P12 2 7 2 . 2 . 1 2 3 .
## MTCO2P12 . 1 . . . . . . . .
## MTATP8P1 . . . . . . . . . .
## MTATP6P1 10 8 . . . . 3 . 2 .There is other important information included in the cm
object. For example, the cells were collected from 19 individuals over
the course of 15 days. Information on which individual and which day
each cell is from is saved within the cm object as column
data.
table(cm$individual)
##
## 18520 18912 19093 18858 18508 18511 18907 18505 19190 18855 19159 18489 18517
## 472 760 925 732 581 701 362 438 524 283 326 400 534
## 18499 18870 19193 19209 19108 19127
## 771 700 373 554 211 353
table(cm$diffday)
##
## day0 day1 day3 day5 day7 day11 day15
## 2383 2389 1691 967 1260 862 448To perform Poisson count splitting we wish to extract only the count matrix.
set.seed(1)
X <- counts(cm)
split <- countsplit(X)## As no overdispersion parameters were provided, Poisson count splitting will be performed.
Xtrain <- split[[1]]
Xtest <- split[[2]]Preprocess the training set.
We now wish to compute clusters on the training set. Unlike in our introductory
tuorial, instead of simply running kmeans() on
log(Xtrain+1), we will use an existing scRNA-seq pipeline
from the scran package that also involves preprocessing
steps such as selecting highly variable genes. In order to do this, we
need to store the training set count matrix Xtrain in a
SingleCellExperiment object.
We could make a new SingleCellExperiment object that
contains only the Xtrain counts matrix. However, this would
discard all of the metadata that was stored in the cm
object, such as information regarding which cells where collected on
which days and from which individuals. Thus, we chose to let
cm.train be a copy of cm where we only update
the counts() attribute. This way, all metadata from
cm is retained in cm.train.
cm.train <- cm
counts(cm.train) <- XtrainNow we are ready for our analysis. These steps were inspired by the
scran
tutorial. We first compute sum factors and then we perform log
normalization. Note that the computeSumFactors() function
requires computing initial clusters of cells using
quickCluster(); these are different from the clusters of
cells that we will later analyze for differential expression.
clusters <- quickCluster(cm.train)
cm.train <- computeSumFactors(cm.train, clusters=clusters)
cm.train <- logNormCounts(cm.train)We then compute the top 2000 highly variable genes. While this step
does not alter the dimension of counts(cm.train), it allows
us to later perform clustering or dimension reduction using only these
highly variable genes.
top.hvgs <- getTopHVGs(modelGeneVar(cm.train), n=2000)Finally, we perform dimension reduction on the dataset (using only the highly variable genes).
cm.train <- fixedPCA(cm.train, subset.row=top.hvgs)Cluster the training set
We cluster the dimension-reduced dataset using scran’s
clusterCells function (which performs graph-based
clustering).
clusters.train <- clusterCells(cm.train,use.dimred="PCA")It turns out that this function returned 11 clusters. We can visualize them below.
table(clusters.train)
ggplot(as_tibble(reducedDim(cm.train)), aes(x=PC1, y=PC2, col=as.factor(clusters.train)))+geom_point()+labs(col="Cluster")## Don't know how to automatically pick scale for object of type
## <reduced.dim.matrix/matrix>. Defaulting to continuous.
## Don't know how to automatically pick scale for object of type
## <reduced.dim.matrix/matrix>. Defaulting to continuous.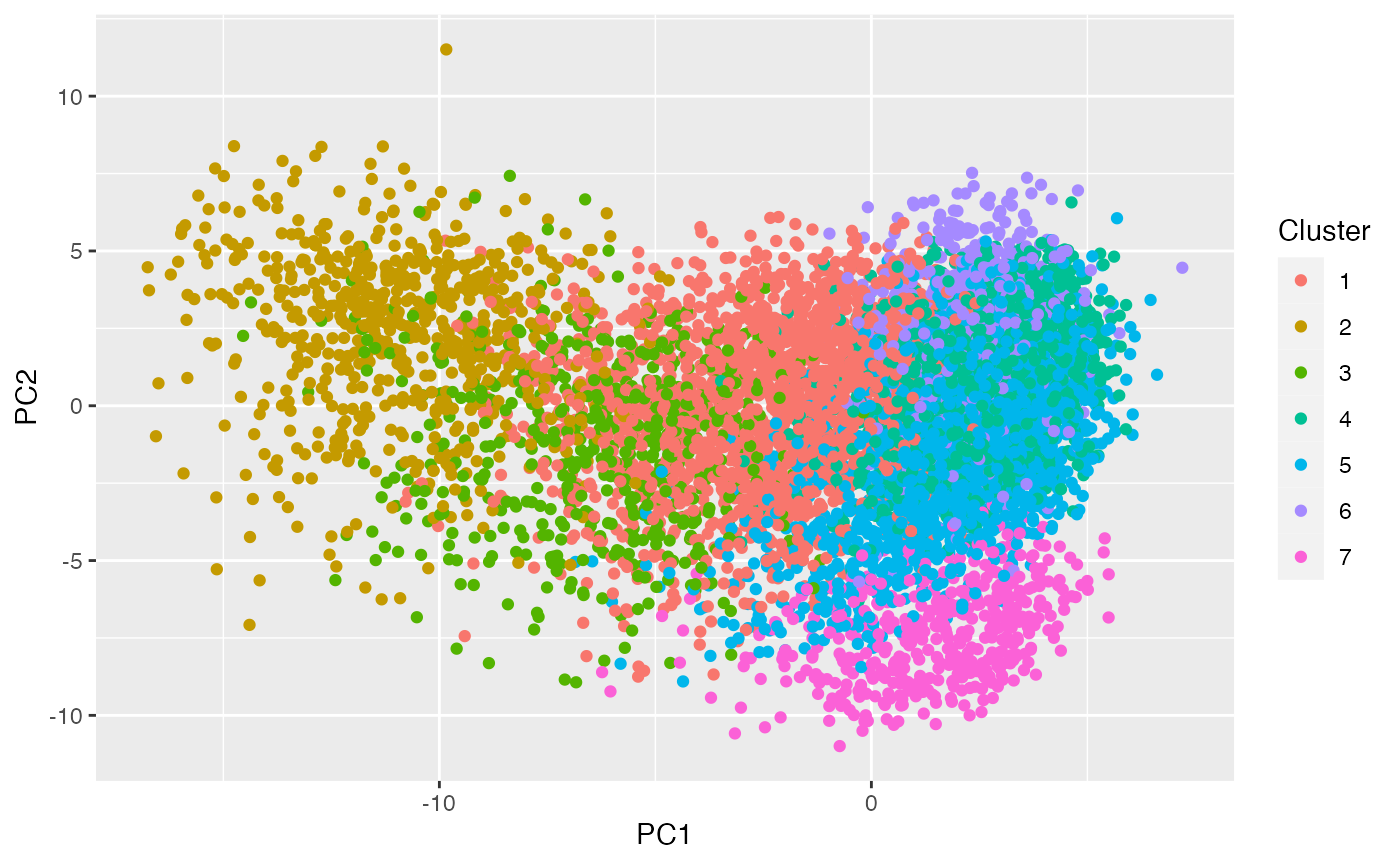
Differential expression testing
Using Poisson GLMs
We now consider fitting Poisson GLMs to test for differential
expression between clusters 1 and 2. We can do this analysis “by hand”,
meaning that we do not need to store Xtest in a
SingleCellExperiment object.
For computational efficiency, we test 500 randomly selected genes for differential expression, rather than checking all 21,000 genes. The first step is to subset the test data to only include these 500 genes and to only include cells that were placed in cluster 1 or cluster 2. We will include size factors for each cell, estimated on the training set, as offsets in our Poisson GLM. We need to obtain the appropriate size factor vector and subset it to only include the cells that were placed in cluster 1 or cluster 2. We do these steps below.
set.seed(1)
indices <- which(clusters.train==1 | clusters.train==2)
genes <- sample(1:NCOL(Xtest), size=500)
Xtestsubset <- Xtest[genes, indices]
sizefactors.subset <- sizeFactors(cm.train)[indices]We are now ready to test for differential expression. We see that 61 out of our 500 genes were identified as significantly differentially expressed at alpha=0.01.
results <- t(apply(Xtestsubset, 1, function(u) summary(glm(u~clusters.train[indices], offset=log(sizefactors.subset), family="poisson"))$coefficients[2,]))
table(results[,4] < 0.01)
##
## FALSE TRUE
## 440 60We can view the names of these highly significant genes below.
head(results[results[,4] < 0.01,], n=10)
## Estimate Std. Error z value Pr(>|z|)
## PPFIA4 1.4853742 0.52704414 2.818311 4.827706e-03
## CDCA7L 0.6667219 0.13899707 4.796662 1.613316e-06
## ZFR 0.2300289 0.08036867 2.862172 4.207492e-03
## SELENOT 0.4791352 0.17424256 2.749817 5.962849e-03
## COL11A2 2.1785214 0.81649008 2.668154 7.626926e-03
## ZNF138 -0.9238206 0.30746879 -3.004599 2.659308e-03
## ZNF436 1.5499127 0.57007870 2.718770 6.552520e-03
## MYL3 4.1843745 0.16801337 24.905009 6.566248e-137
## EDEM3 0.5632183 0.21669065 2.599181 9.344635e-03
## MTUS1 1.3821900 0.31984429 4.321446 1.550097e-05Using scran
In this section, instead of testing for differential expression “by
hand” using glm(), we use the scoreMarkers()
function from the scran package. This is the method used in the scran
tutorial, in the section titled “Find Marker Genes”.
In order to use the scoreMarkers() function, we need to
store our test matrix in a SingleCellExperiment object. We
can either construct this from scratch using only the count matrix, or
we could make a copy of the original cm object and add the
count matrix after. Since we do not anticipate needing any metadata in
this section, we construct the object from scratch.
cm.test <- SingleCellExperiment(list(counts=Xtest))## Warning in S4Vectors:::anyMissing(runValue(x_seqnames)): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")## Warning in S4Vectors:::anyMissing(runValue(strand(x))): 'S4Vectors:::anyMissing()' is deprecated.
## Use 'anyNA()' instead.
## See help("Deprecated")The scoreMarkers() function needs access to the
log-normalized counts assay (logcounts()) in the
cm.test object. To fill in this assay, we need to run the
logNormCounts function on cm.test. We would
like to normalize the test matrix using size factors computed on the
training set (this follows our general philosophy of performing all
preprocessing steps on the training set). Thus, we obtain size factors
from the training set before calling logNormCounts().
sizeFactors(cm.test) <- sizeFactors(cm.train)
cm.test <- logNormCounts(cm.test) This first element in results shows marker genes that
distinguish cluster 1 from all other clusters.
results <- scran::findMarkers(
cm.test, groups= clusters.train,
pval.type = "all")
results[[1]]## DataFrame with 21971 rows and 9 columns
## p.value FDR summary.logFC logFC.2 logFC.3
## <numeric> <numeric> <numeric> <numeric> <numeric>
## TPM1 5.14786e-85 1.13104e-80 1.5604602 -2.526001 1.5758556
## NEXN 9.79295e-23 1.07580e-18 -0.6131618 -1.221489 0.1960131
## KRT18 8.76435e-18 6.41872e-14 0.3717481 0.371748 0.5273134
## ACTA2 2.12218e-17 1.16566e-13 0.0923831 -0.546589 0.0954392
## ANKRD1 4.04560e-17 1.77772e-13 0.2718263 -1.504769 0.3379046
## ... ... ... ... ... ...
## ABCD1 1 1 0 -0.00727795 -0.000768208
## CLIC2 1 1 0 0.00000000 0.000000000
## EEF1A1P41 1 1 0 -0.00408911 0.000000000
## PCDH11Y 1 1 0 0.00000000 -0.001357497
## TMSB4Y 1 1 0 0.00000000 -0.000286135
## logFC.4 logFC.5 logFC.6 logFC.7
## <numeric> <numeric> <numeric> <numeric>
## TPM1 1.3627284 1.7967622 -1.873952 1.560460
## NEXN 0.1688780 0.1950033 -0.613162 0.205480
## KRT18 0.4117986 0.5686877 -0.729451 0.580191
## ACTA2 0.0949788 0.0923831 -1.156074 0.101023
## ANKRD1 0.3082924 0.3164633 -1.559440 0.271826
## ... ... ... ... ...
## ABCD1 -0.001191767 -0.002529505 0.00000000 -0.00339277
## CLIC2 -0.001037117 -0.002346766 -0.00617463 0.00000000
## EEF1A1P41 -0.000278101 0.000000000 0.00000000 0.00000000
## PCDH11Y -0.001090408 -0.000844056 0.00000000 0.00000000
## TMSB4Y 0.000000000 0.000000000 0.00000000 -0.00672774Even with count splitting, we identify several highly significant
marker genes, both “by hand” and with the findMarkers()
function.